When studying cancer or any disease one of the things we are interested in are the alterations that cause the disease. By now, we have quite some arsenal to study genomic aberrations, however assigning those to a specific phenotype is not trivial. This is particularly true for changes affecting metabolism, since there is a myriad of regulation events that take place after gene expression and which drive metabolism.
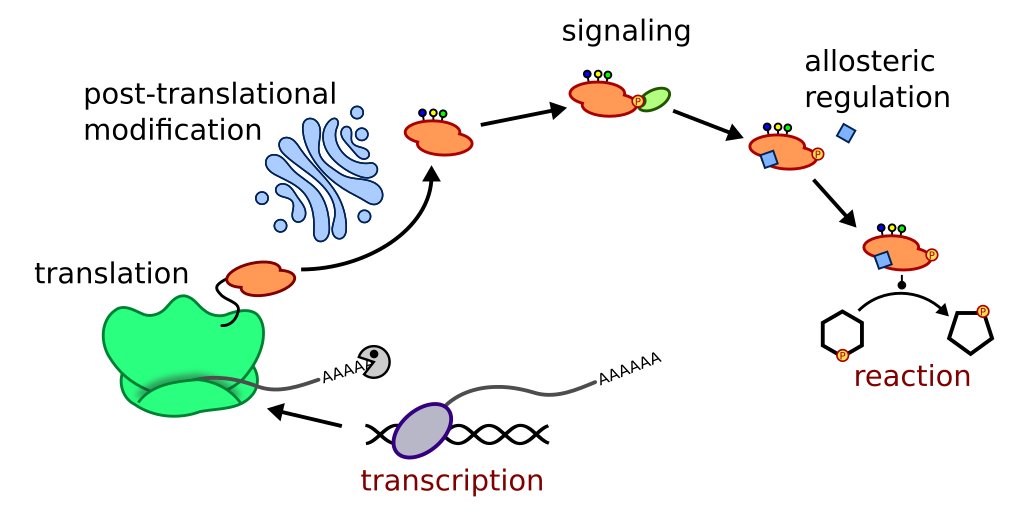
The image above shows just an example of events that can happen between translation of an enzyme gene until it will finally catalyze a reaction. Metabolome data is naturally closer to the phenotype, however it usually does not give us information on which alterations caused the observed phenotype.
In our recent paper we introduce a method that identifies potential alterations in enzymatic activity that explain the observed metabolome data by a combination of simple statistics and metabolic modeling. Here, we employ the k-cone, the space of all feasible enzyme reaction rates. Identifying changes in the k-cone and restricting the resulting set to those that are not expected to vary a lot in normal conditions we obtained enzymes with altered kinetics in the HeLa cancer cell line. Here, the data we used was obtained by measuring metabolite concentrations for some 40 metabolites in HeLa and HaCaT cells.
We showed that the obtained altered enzymes are well-known drivers of cancer progression and that those alterations do not correlate with gene expression data, indicating that many of them are caused by post-translational regulation.
The analysis is implemented in an open source package for R called dycone.