In those days analyzing microbiome data often means dealing with read counts from high-throughput sequencing. Usually, we stratify those counts by some entity of interest: phyla, species, genes, sequence variants and so on. Three questions that may pop up when analyzing those read counts are the following:
- Does the sample contain enough reads to observe the majority of phyla, species, genes that are actually in the sample?
- How well do the reads represent their distribution in the sample?
- How do I compare different samples with strongly varying total read counts?
Let’s assume we ran a metagenomics experiment and obtained read counts for a lot of genes across many samples. One thing that might interest us is the proportion of the genes in the samples. One thing we could do to alleviate the problem of different library sizes, e.g. different total read counts, would be to divide the gene-level counts by the total counts for each sample which basically gives us an estimation of the discrete probability distribution of genes in the sample. That distribution can be seen as the probability of observing a particular gene if I would generate one additional read in my experiment. However, that creates some problems. For instance if there are 100,000 genes in total and I only have 10,000 reads, there is no way to observe a read for each gene. So if we get the proportions by dividing by the total read counts we will make two principal errors:
- We will underestimate the probability of observing a new gene since we assign a probability of 0 to that event, because assume that the observed genes are the only genes in the sample.
- We will overestimate the probability of the observed genes, since the probabilities sum to one and we made error #1. So basically we distribute the entire “probability mass” across the observed genes, but forget about the unobserved ones.
In summary, overestimation of common events is often associated with the underestimation of uncommon ones.
Often we find two recommendations to deal with the problem:
- rarefaction, meaning to subsample all of the samples to have the same total read count (the smallest observed one)
- some kind of pseudo-counting which adds a constant to all counts and estimates probabilities based on that. This one also assigns a count of one to the “new” gene event.
Getting to a real example
To study the effect of rarefactions and pseudo-counts on a more relevant example, let us get a real world example. You can find all the data we use here on figshare. We will use gene read counts for a swedish healthy individual (SRA sample ERR260132). We will not go into too much details how we got to those counts from the raw data (another blog post? ^_^’). In brief: reads were trimmed and filtered aligned against the IGC genecatalogue and gene-level counts extracted with RSEM. This leaves us with counts for about 660,000 genes (IGC contains a little less than 10 million genes) across 10 million reads. For a lack of better knowledge we will assume the those counts represent the “real” discrete distribution and sample counts from that distribution (which is what rarefaction does).
First thing we will need is to load the the data and get the dicrete distribution. I have provided a csv with the counts for us, so that is pretty straight forward.
|
|
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 9.630e-08 9.630e-08 2.889e-07 1.479e-06 8.668e-07 1.708e-03
So our discrete probabilities are now in stored in real. We see there some pretty common and also pretty uncommon genes.
We also need a function to sample a new count vector from that distribution.
|
|
We use tabulate here since it will be much faster than the regular table, albeit using a bit more memory.
To perform our benchmark we still need a quantity to benchmark. We could just get the mean squared or absolute distance of the sample probabilities to the “real” ones (sometime called “histogram distance”), however that is often not what we are interested in. For instance, if the probability for a gene is 0.5 an error of 0.001 seems pretty small, but if the probability for a gene is 0.0001 an error of 0.001 is pretty high (10 times larger than the actual probability). So we might prefer something relative. If we denote the real probability for a gene as p(g) and our estimate as q(g) the following might be more interesting:
$$ error = \frac{p(g) - q(g)}{p(g)} = 1 - \frac{g(g)}{p(g)} $$
This gives us the fractional error. We can drop the constant 1 and simply quantify the quotients of the estimated and “real” probabilities (1 being the best value). We will summarize the values across all observed genes using the median.
|
|
So what does that give us for our data set?
|
|
## Need help getting started? Try the cookbook for R:
## http://www.cookbook-r.com/Graphs/
|
|
## `geom_smooth()` using method = 'loess'
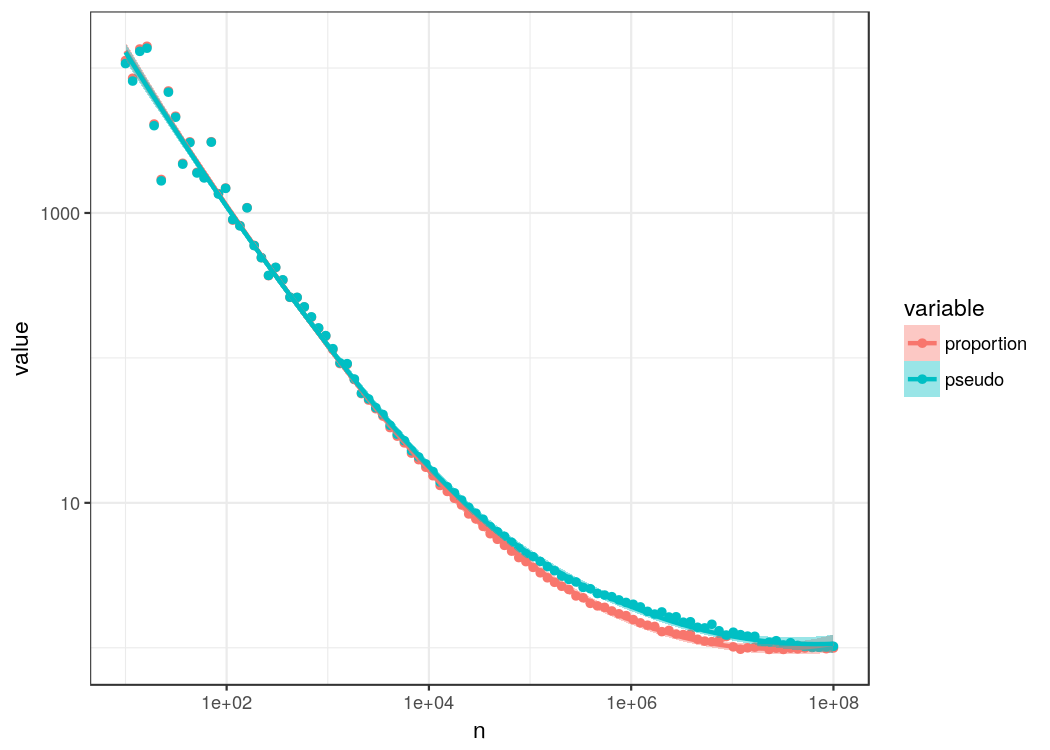
First obvious thing we see is that subsampling, which is the same thing done in rarefaction, really messes with the estimation. In median we are overestimating probabilities by a lot. So if you are interested in getting a good estimation of proportions/probabilities, rarefaction is not the best choice. In fact, even if you are only interested in relative changes between samples there are almost always better strategies as discussed in an excellenct article by Susan Holmes’ group. Simple proportions and pseudo counts basically perform the same. Actually, proportions have a pretty bad rep in microbiome research. They are an unbiased estimator and the maximum-likelihood estimate and have decent convergence. It is more a problem of sample sizes than the problem of a bad estimator. There are many alternative methods to estimate probabilities based on event counts, however many requires some kind of additional assumptions or prior distributions (when using Bayesian methods). Is there something simple we could try that does not require additional information?
What does the Enigma have to do with the microbiome?
The problem of getting probabilities from observed events is obviously much older than microbiome research. It has arisen in language analysis where one is usually interested in the probabilities of words given only text excerpts. One particular critical situation where the problem came up was doing the deciphering of the Enigma messages. Here Good and Turing had to come up with good probability estimates for rare messages from small sample sizes since waiting for a lot of messages might have dire consequences. Even though only preserved in formula, the resulting Good-Turing estimator and its derivatives would dominate in language analysis for a long time after World War II. The theory behind their estimator was taken up by Orlitsky and colleagues in 2003 who wanted to understand why it performed better for rare events than other classical estimators. Unfortunately, Turing’s own intuitive explanantion of the estimator was not preserved.
After the war, Good published the estimator (7), mentioning that Turing had an “intuitive demonstration” for it but not describing what this intuition was [from Orlitsky’s paper]
It revolves around two basic quantities, multiplicity which is how often an event occured in a series and prevalence which is how often a particular multiplicity occurs (the count of counts or multiplicity of the multiplicity :D). For instance if we have a count of one for many events (many uncommon event) we could expect the probability for a new event to be high. In their remarkable work Orlitsky et. al. defined the concept of attenuation which is related to the quotients between the maximum probability pmax of an event under any distribution (including the real one) and a probability estimator q (so pmax/q). In essence this is related to underestimation of probabilities since pmax/q >= 1 implies that one never underestimates a probability. They showed that any pseudo count method has asymptotically unlimited attenuation and that the Good-Turing estimator has asymptotocally constant attenuation larger than one. Maybe the most important part of their work was to come up with a set “diminishing attenuation” estimators that asymptotically converge to an attenuation of 1, meaning that as the sample size gets larger the estimator tends to never underestimate the probability of an event.
So what does that mean for us? Diminishing attenuation estimators are great if we want to avoid underestimation, however, we observed mostly overestimation. But: we also argued earlier that overestimation is associated with underestimation of the probability of unobserved (new) events which is a problem the diminishing attenuation estimator solves. In particular attenuation is closely related to our error measure (p_max/q vs q/p_real). So we should certainly try it out…
Here, we will use the q2/3 estimator that approaches an attenuation of 1 with a rate of
$$2^{O(\sqrt[3]{n})}$$
There is also a q1/2 but it has super-polynomial complexity. The q2/3 method has linear complexity and optimizing a bit for fast calculation we end up with the following:
|
|
This method will assign a probability to each observed event, but those will not sum to one since we also consider the probability for a “new” event which is given by p(new) = 1 - sum(probs).
Updating our benchmark and also tracking the estimated probability for a new event we get the following:
|
|
## `geom_smooth()` using method = 'loess'
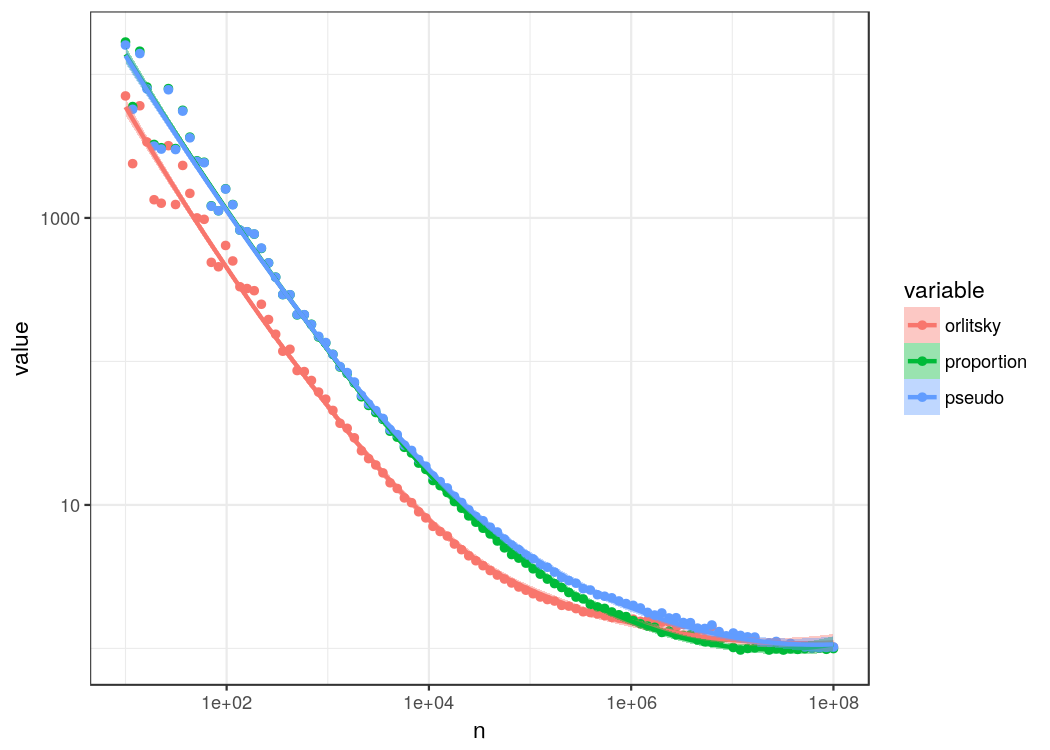
Well, it seems to work. Relative errors are certainly smaller with Orlitsky’s diminishing attenuation estimator. We can also see that it draws with the other estimators around a n of one million which is where we expect to have seen most of the actual events, so the advantage of estimating the probability for an unobserved event is lost. The probability estimate for observing a new event can be substantial and is larger 50% for the first few n.
What about taxonomy levels?
Not all of us might be interested in gene counts. Often we are more interested in the taxonomy, for instance species in the sample. Let’s quickly look at another data set (ERR414335) which comes from a danish patient with Type 1 Diabetes. Here, the species were quantified using SLIMM. Again, the data is presented as CSV and contains read counts for 102 species in the sample and one unknown category for reads that could not be aligned uniquely to a single species.
|
|
## `geom_smooth()` using method = 'loess'
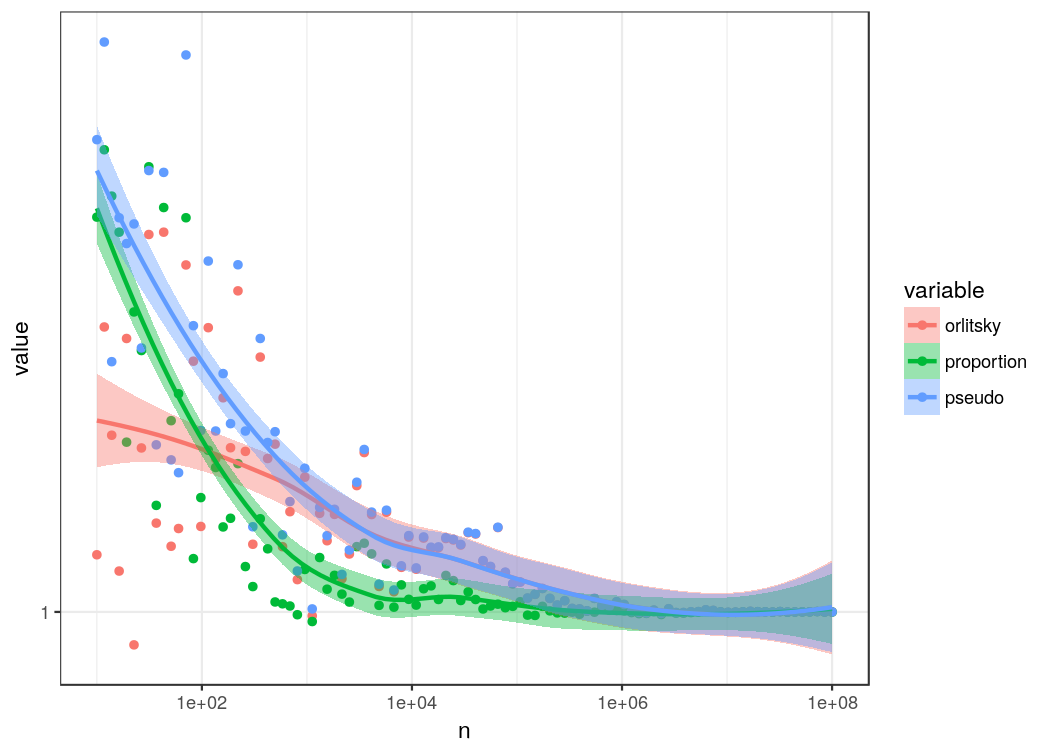
In general, there is a lower level of overestimation. Again, the diminishing attenuation estimator loses its advantage after passing the number of discrete events (species in this case). You might ask yourself why the estimator seems to overestimate probabilities more than the proportion estimator. This, is due to the fact that Orlitsky’s estimator assigns a probability close to p_max which is not necessarily the “real” distribution. In particular it still considers that there might be unobserved events which is probably correct. In the end our “real” distribution was taken from a population sample.
Summary
So in general we have seen that diminishing attenuation estimators give some advantage over classical proportion based methods if the sample size is low. This advantage arises particularly when your principal error source is underestimating the occurrence of new genes, species, etc. Most estimators seem to perform pretty well after the sample size approaches the number of categories. In practice we probably do not know that number. So, we are back to the problem of judging whether our sample size was sufficiently large for the particular question we want to ask. In my opinion, the really amazing part of the diminishing attenuation estimator is the probability estimate for the “new” event, not the estimates for the observed events. The probability estimate for new events from the diminishing attenuation estimator is an approximate upper bound of the probability to observe a new event when increasing the sample size by one read. For instance for the gene count data set this probability is 0.002 and for the species count data set it is 3.5e-7. This gives us a clear quantification of how sure we can be that we have sampled enough which I feel may be pretty useful.
Also, if you are interested in a much more complete overview of what Good and Turing have done for Science I highly recommend the linked lecture of Susan Holmes.
Last but not least, if you have another estimator you would like me to try with the presented data sets do not hesitate to contact me on twitter or by mail and I will happily add analyses for those.
